Deutsch
Benutzerdefinierte Teilefertigungslösungen
Medizinische Geräte Teilefertigung
Neway ist auf die Herstellung von medizinischen Gerätekomponenten spezialisiert und bietet CNC-Bearbeitung, 3D-Druck, Vakuumguss, Druckguss und Spritzgießdienstleistungen an. Wir stellen sicher, dass wir hochpräzise und biokompatible Komponenten liefern, die den strengen Branchenstandards entsprechen und zuverlässige sowie langlebige Lösungen für die medizinische Geräteindustrie bieten.
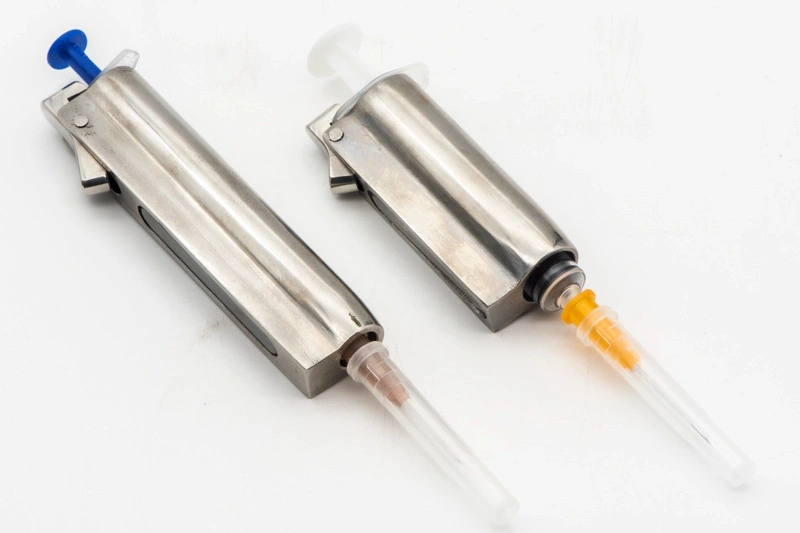
Medizinische Geräte Teilebearbeitung
Die medizinische Gerätebearbeitung umfasst präzise CNC-Bearbeitungsverfahren wie Fräsen, Drehen, Bohren und Schleifen, um qualitativ hochwertige Komponenten für medizinische Anwendungen zu produzieren. Mit Multi-Achsen- und Präzisionsbearbeitungstechniken sowie Funken-Erosion-Bearbeitung (EDM) werden enge Toleranzen und Zuverlässigkeit gewährleistet. Diese Verfahren sind entscheidend für die Herstellung von Teilen, die den strengen Sicherheits- und Leistungsstandards der Medizinbranche entsprechen.

Materialauswahl für Medizinische Geräte
Materialien wie Superlegierungen, Titan, Aluminium, Kupfer, Messing, Bronze, Kohlenstoffstahl, Edelstahl, Kunststoffe und Keramiken sind entscheidend in der Herstellung medizinischer Geräte. Diese Materialien bieten Haltbarkeit, Biokompatibilität und Präzision für Implantate, chirurgische Instrumente und medizinische Geräte.

Typische Oberflächenbehandlungen für medizinische Geräte Teile
Typische Oberflächenbehandlungen für medizinische Geräte Teile umfassen Anodisieren, Elektropolieren, PVD, Pulverbeschichtung, Passivierung und Wärmebehandlung. Diese Behandlungen verbessern die Biokompatibilität, Korrosionsbeständigkeit und Haltbarkeit. Prozesse wie Elektropolieren und Anodisieren verbessern die Oberflächenstruktur, während Beschichtungen wie Teflon oder UV-Beschichtungen zusätzlichen Schutz bieten, um sicherzustellen, dass medizinische Geräte die strengen Leistungs- und Sicherheitsstandards erfüllen.

Erfahren Sie mehr
Thermal Coating

Erfahren Sie mehr
As Machined

Erfahren Sie mehr
Painting

Erfahren Sie mehr
PVD (Physical Vapor Deposition)

Erfahren Sie mehr
Sandblasting

Erfahren Sie mehr
Electroplating

Erfahren Sie mehr
Polishing

Erfahren Sie mehr
Anodizing

Erfahren Sie mehr
Powder Coating

Erfahren Sie mehr
Electropolishing

Erfahren Sie mehr
Passivation

Erfahren Sie mehr
Brushing

Erfahren Sie mehr
Black Oxide

Erfahren Sie mehr
Heat Treatment

Erfahren Sie mehr
Thermal Barrier Coating (TBC)

Erfahren Sie mehr
Tumbling

Erfahren Sie mehr
Alodine

Erfahren Sie mehr
Chrome Plating

Erfahren Sie mehr
Phosphating

Erfahren Sie mehr
Nitriding

Erfahren Sie mehr
Galvanizing

Erfahren Sie mehr
UV Coating

Erfahren Sie mehr
Lacquer Coating

Erfahren Sie mehr
Teflon Coating
CNC-Bearbeitungslösungen für medizinische Geräte
CNC-Bearbeitung ist entscheidend für medizinische Geräte zur Herstellung präziser Teile wie chirurgischer Instrumente, Implantate und Diagnosegeräte, die strenge Anforderungen an Präzision, Biokompatibilität und Sicherheit erfüllen.








Starten Sie noch heute ein neues Projekt
Designrichtlinien für medizinische Geräte Teile
Medizinische Geräte Teile erfordern Präzision, Sauberkeit, Vorschriften und Sicherheit für den Benutzer. Dieser Artikel umreißt die ingenieurtechnischen Prinzipien, um sicherzustellen, dass die Teile klinisch funktionsfähig, leicht zu reinigen und zertifiziert sind.
Frequently Asked Questions
Verwandte Ressourcen erkunden



Neway Precision Works Ltd.
Nr. 3 Lefushan Industrie-Weststraße
Fenggang, Dongguan, China
PLZ 523000
Lösungen
Copyright © 2026 Machining Precision Works Ltd.All Rights Reserved.